제약사의 투명한 데이터 관리는 데이터 완전성 측면에서 매우 중요하다.
특히 시장에 유통될 의약품은 투명한 환경에서 적절히 제조되었다는 근거가 있어야만 유통될 수 있다.
그렇지 않았을 경우, 어떤 교차오염의 위험이 있을지, 예측하지 못하는 불순물이 섞여 문제를 일으킬지 모르기 때문이다.
Tianjin Darentang Jingwanhong Pharmaceutical Co., Ltd.는 중국 톈진시에 위치한 제약회사로, 전통 한약을 기반으로 한 다양한 의약품을 생산하고 있다. 1980년대에 설립되었으며, 중국 명나라 시대로 거슬러 올라가는 유서 깊은 약국인 'Lejia Laopu'의 전통을 계승하고 있다.
최근 이슈가 된 FDA의 텐진제약에 보낸 warning letter를 분석해보겠다.
FDA는 2024년 11월 24일 이를 공개했고 3월 18~22일 FDA 조사관의 실사 현장 접근을 제한하거나 거부했다고 설명했습니다. 이에 해당 시설에서 제조·처리·포장 또는 보관한 제품에 대한 cGMP 규정을 위반해 불완전한 환경에서 제조한 것으로 간주한다고 덧붙였다.
원문을 보면,
This warning letter summarizes significant violations of Current Good Manufacturing Practice (CGMP) regulations for finished pharmaceuticals. See Title 21 Code of Federal Regulations (CFR), parts 210 and 211 (21 CFR parts 210 and 211).
이 significant violation을 세가지로 나눌 수 있습니다.
1)Failure to clean, maintain, and sanitize equipment → 장비 청소 및 유지보수 미흡 (21 CFR 211.67(a))
In your response, you acknowledge the incomplete cleaning of filling machine ID-192 and state that the equipment’s cleaning procedure is revised. You also state that the conditions identified above filling machine ID-197 are repaired.
Your response is inadequate because you did not implement sufficient corrective actions related to the status and cleaning of all equipment used in the manufacture of drugs intended for the U.S. market. You also failed to evaluate the potential impact of your filling machines’ condition and inadequate cleaning on the quality of your distributed drug batches. (warning letter 일부 발췌.)
이에 대해 텐진제약에서는 문제가 되는 filling machine에 대해 청소 과정을 다시 고안하였고 수리하였다고 답변했다.
하지만 FDA에서는 이를 부적절하다 판단하였다. 미국 시장에 유통될 의약품의 제조에 있어서 모든 장비에 대한 청소가 충분한 Corrective Action이 아니다, 또 잠재적인 영향을 고려하지 않았다는 근거를 제시하였다.
해당 문제에 대하여 FDA가 제안한 내용
1) CAPA(시정 및 예방 조치) 계획 수립
설비 및 장비의 운영 관리를 철저히 감시할 수 있도록 CAPA 계획을 마련해야 함.
설비/장비 성능 문제의 신속한 감지
효과적인 수리 조치 수행
적절한 예방 유지보수 일정 준수
설비 및 시설 인프라의 기술적 업그레이드
지속적인 관리 검토 시스템 개선
2) 청소 효과성에 대한 독립적인 평가
교차 오염 위험성을 평가하기 위한 포괄적이고 독립적인 검토 필요.
주요 내용:
남아 있는 잔류물 확인
부적절하게 청소된 다른 제조 장비 조사
교차 오염된 제품이 유통되었는지 평가
청소 절차 및 실행상의 문제점 도출
3) 청소 프로그램 개선을 위한 CAPA 계획
위 평가를 바탕으로 청소 절차 및 실행 방안을 보완하는 CAPA 계획 수립.
주요 내용:
장비 청소의 취약점 분석 및 개선
청소 효과성 강화
모든 제품과 장비에 대한 청소 검증 절차 개선
기타 필요한 보완 조치 및 실행 일정 포함
2) Failure to establish adequated written produres
Your firm failed to establish adequate written procedures for production and process control designed to assure that the drug products you manufacture have the identity, strength, quality, and purity they purport or are represented to possess (21 CFR 211.100(a)).
Based on the limitation of the inspection described above, you failed to provide data to demonstrate that you adequately validated your manufacturing processes used to manufacture your OTC drug product and to demonstrate that your processes are reproducible and controlled to consistently yield drugs of uniform character and quality.
Although you state your manufacturing process is validated and equipment is qualified, you do not submit supporting information.
In response to this letter, provide:
• A complete and unredacted process validation protocol and report for your drug product “(b)(4).”
• A complete and unredacted batch record for the most recent batch of “(b)(4)” released and shipped to the United States for each drug product configuration (i.e., (b)(4)g, (b)(4)g, and (b)(4)g).
• A complete and unredacted qualification report for the following manufacturing equipment: (b)(4) tank ID-447, (b)(4) tank ID-344, and (b)(4) tank ID-264.
결론적으로 완전한 process validation protocol과 report를 갖춰야 한다. (검열되지 않은 데이터를 기록하여야함)
배치 기록과 최근에 시장에 출하된 배치에 대한 기록, 그리고 제조 시설에 대한 qualification report.
결국 텐진 제약이 출하한 배치에 대하여 신뢰를 제공할 수 없다는 것이다. 기록을 검열하거나, 삭제하면 안된다.
또한 시설에 대한 적격성 레포트를 제공하여야 한다.
3) You firm failed to establish required laboratory control mechanisms (21 CFR 211.160(a)), including those related to stability studies (21 CFR 211.166).
Also, your firm has not performed forced degradation studies to identify degradants that may be present in sufficient quantities to require testing during stability studies.
In your response, you commit to conduct an “impact factor” and accelerated test to assess the impact of different factors on your drug product stability. You also committed to test three batches per production schedule.
Your response is inadequate because you did not provide details of your “impact factor” and accelerated test plan and how they relate to the lack of degradation studies. Your plan also fails to include a retrospective evaluation of batches that have been released and are currently within expiry in the U.S. market.
수행하여야 하는 forced degradation study 즉, 강제 분해 실험을 진행하지 않았다. 이는 안정성시험중에 생길 수 있는 분해산물을 식별하기 위함인데 이를 수행하지 않았다는 것은 큰 문제가 된다. 분해산물을 명확히 알 수 없는 제품을 어떻게 시장에 내놓을 수 있을까?
텐진제약은 이에 대한 답변으로 impact factor를 유도하고 의약품의 안정성에 영향을 줄 수 있는 다른 요인을 평가하기 위해 가속화시험을 했다고 주장했다. 하지만 이 impact factor라는 게 정확하게 무엇인지, 그리고 가속화 시험이 이 분해 실험을 하지 않은 것과 무슨 관련이 있는지 충분히 설명되지 않았다. 그리고 회고적 평가 또한 제공하지 않았다.
• A report for the “impact factor” and accelerated tests for the analytical method for U.S. drug product. Include details of any changes implemented and how the results may impact previously analyzed batches.
이 import factor라는 것 에 대한 report, 가속화 시험에 대한 report가 필요하다. 그 결과가 이전에 분석된 배치에 어떤 관련이 있을지 제공해야 한다.
• A comprehensive assessment and CAPA plan to ensure the adequacy of your stability program. Your remediated program should include, but not be limited to:
o Stability indicating methods.
o Stability studies for each drug product in its marketed container-closure system before distribution is permitted.
o an ongoing program in which representative batches of each product are added each year to the program to determine if the shelf-life claim remains valid.
o detailed definition of the specific attributes to be tested at each station (timepoint)
o all procedures that describe these and other elements of your remediated stability program.
안정성 프로그램의 적절함을 보장하기 위한 평가, CAPA 계획을 제공해야한다. 용기-마개 시스템상에서의 안정성 시험, 그리고 등등등...
• A list of chemical and microbial specifications, including test methods, used to analyze each batch of your drug products before a disposition decision.
o An action plan and timelines for conducting full chemical and microbiological testing of retain samples to determine the quality of all batches of drug product distributed to the United States that are within expiry as of the date of this letter.
o A summary of all results obtained from testing retain samples from each batch. If such testing reveals substandard quality drug products, take rapid corrective actions, such as notifying customers and product recalls.
화학물질, 미생물 리스트, 시험 방법 리스트가 필요하다.
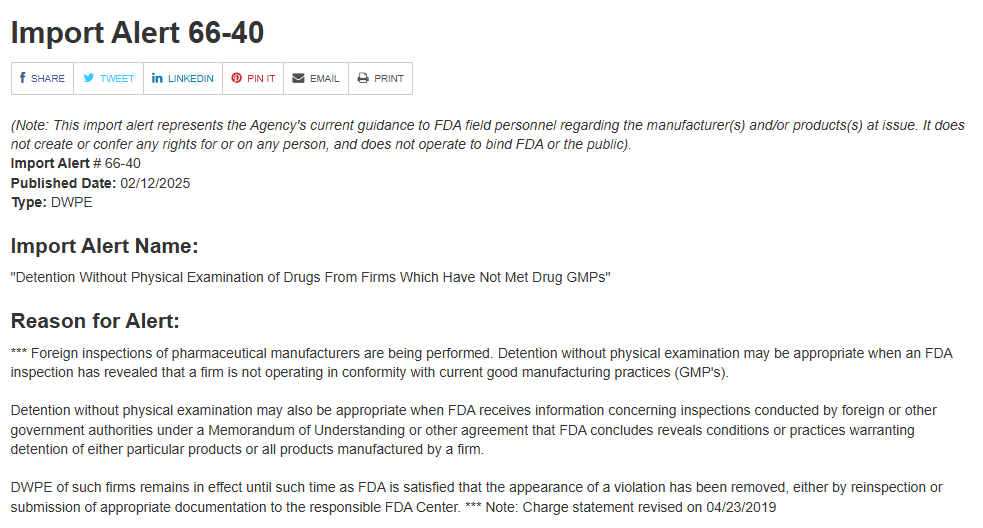
FDA는 텐진 제약에 import alert 66-40을 암시했다. 이는 회사가 cGMP를 제대로 지키지 않다는 것을 조사를 통해 밝혀냈을 때 의약품의 물리적인 조사 없이도 detention을 진행하는 것이다. 여기서 detention은 압류의 의미로 쓰였다.
결론적으로 cGMP를 잘 지키기 위해서는 의약품을 제조할 때 제조하는 것 뿐 아니라 청소 과정, 시설 관리에 신경을 써야한다. 단순히 제조만 잘 하는 것을 넘어서 잠재적인 위험 요소를 관리하고 항상 check - corrective action + predictive action까지 가능하게.. 또한 배치에 대한 기록은 늘 투명하게 관리하여야한다. 시험 과정도 의약품의 안정성을 보장할 수 있는 시험인지 검증해야 한다.
Tianjin Darentang Jingwanhong Pharmaceutical Co., Ltd. - 683619 - 10/30/2024 | FDA
Tianjin Darentang Jingwanhong Pharmaceutical Co., Ltd. - 683619 - 10/30/2024
CGMP/Finished Pharmaceuticals/Adulterated
www.fda.gov